Research
While it has long been assumed that evolution is a slow and gradual process, recent studies show that evolution can in fact be rapid and occur on human-relevant and ecological timescales. For example, in natural populations of insects, pesticide resistance can evolve within a mere decade, and in host gut microbiomes, antibiotic resistance can evolve within just a matter of just 1 day. Given the profound impacts of global change and rapidly fluctuating selective pressures on natural diversity and public health, rapid evolution is a force that is needed to be both understood as well as exploited. Our research group is focused on understanding the dynamics of evolution and its intersection with ecological processes in natural populations exposed to shifting selective pressures.
Our group has been exploring these dynamics in multiple model systems. Below, we describe our work in human microbiomes and eukaryotes.
Evolutionary dynamics in the human microbiome
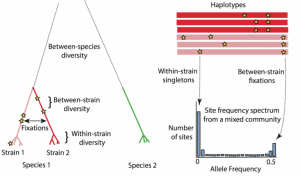
In our recent work, we quantified the evolutionary dynamics of roughly 40 prevalent species of gut bacteria. We found that gut bacteria can evolve in humans in the span of just days to months. While several of these adaptations are transient, some can spread from host to host, indicating that there are shared selective pressures across hosts.
Simply uncovering evidence for evolution in the microbiome is just the start to fully characterizing the extent and limitations of evolution in this complex community. With a bounty of genetic variation, evolutionary inquiry in the human microbiome is an exciting frontier for future research, with important biomedical implications. Some of the questions we are pursuing include:
- How rapid is evolution within hosts?
- Is selection across a population of hosts common?
- How does the community composition constrain or accelerate evolution in the microbiome?
- How does space and rates of migration impact rates and modes of adaptation?
- Are soft sweeps common in the microbiome?
- How do deleterious variants impact adaptation in the microbiome?
Rapid adaptation in Eukaryotic populations
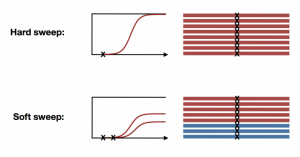
With the availability of fully sequenced whole genome data, especially from natural populations, many of our assumptions about molecular evolution have been challenged. For example, it has long been assumed that natural selection can be slow and infrequent, resulting in signatures of hard sweeps. We developed haplotype statistics to detect and classify hard and soft selective sweeps in fully sequenced whole genomes. Applying these statistics to natural populations of D. melanogaster revealed abundant soft selective sweeps on autosomes, suggesting that adaptation can in fact be rapid. However, on the X chromosome, we find evidence for elevated rates of hard sweeps, potentially due to lower effective population size or hemizygosity.
Several questions remain based on this work, including:
- Why are partial sweeps so common?
- Is there a demographic model that fits multiple summary statistics including LD and pi from Drosophila population genomic data?
- How do we detect selection from from non model organism data, where sample sizes are shallow, or sequencing quality is poor?