Gene-specific selective sweeps are pervasive across human gut microbiomes
We are delighted to share that our manuscript has been recently published in Nature.


We are delighted to share that our manuscript has been recently published in Nature.

Thrilled to share the news that Adam Park is a finalist for the Simons Eco Evo graduate fellowship! Adam joins us from Penn State University where he completed his MS in CS and Math. He recently wrote a paper on k-mer sizes, which can be viewed here.

Aina Martinez i Zurita, Peter Laurin, and Yashas Appaji participated in a 2 minute grad slam at the annual QCBIO retreat and won best talk prize!


Congratulations to Ricky Wolff for successful completion of his PhD! In his whirlwind PhD, Ricky discovered a number of important facets of the human gut microbiome. In his first paper, Ricky discovered that strains in the human gut are stable through time, and that they display ecological characteristic of species-level diversity (Wolff et al. mBio 2023). Subsequently, he discovered that gene-specific selective sweeps are pervasive across human gut microbiomes, with an enrichment for adaptations at carbohydrate-digestion related loci and clear differences in selective pressures in different parts of the world with different lifestyles (now accepted at Nature!). Along the way, he made many seminal contributions to other papers in the lab, including showing that the diversity begets diversity hypothesis extends to the strain level, that strains show uniform frequencies along the gut, and that in fruit flies there are genetic elements important for colonization of strains. We are very excited for Ricky’s next steps as a postdoc at UCSD!


Postdoc Maya Weissman was selected to be a QCBIO collaboratory fellow! This stipend supports her not only in her research, but also provides a teaching opportunity to teach a workshop to the broader UCLA community this fall. Maya will be conducting a workshop on running population genetic simulations in SLiM!

Congratulations to Michael Wasney for winning best talk prize at the recent microbiome virtual international forum! He presented on his recent paper on Uniform Genetic Diversity Along the Tract of the Gut.
Watch his talk here.

Congratulations to Dr. Mariana Harris for her successful PhD defense! In her highly productive PhD, Mariana wrote a number of papers on the mode and tempo of natural selection in Drosophila and ancient humans. In her first two papers, Harris et al. 2023 MBE and Harris et al. 2024 Genetics, Mariana showed that the X chromosome harbors more hard sweeps than autosomes in multiple species of Drosophila. In her latter two papers, she showed that hard selective sweeps are common in modern and ancient humans and that sweeps were both lost and persisted in the face of human admixture (Pandey*, Harris* et al. 2024 Nature Communications; Harris et al. 2025 bioRxiv). We are really excited for her next steps!



We are delighted to welcome postdoctoral scholar Maya Weissman to our group. She is joining us after having successfully defended her PhD on bet hedging at Brown University with Prof. Dan Weinreich. Welcome Maya!

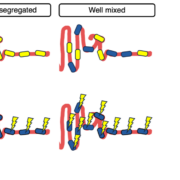
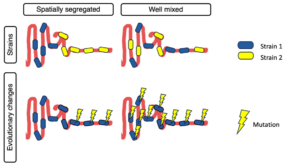
We are delighted to share our latest manuscript on the distribution of genetic diversity along the gut. In this work, we analyze the luminal contents of mice inoculated with the same human stool sample and find to our surprise that genetic diversity is uniform along the gut, rather than spatially segregated as observed at the species level.
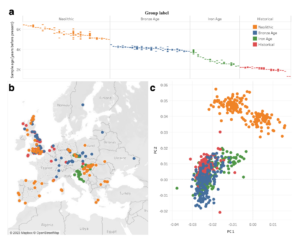
We are happy to share that our manuscript on Leveraging ancient DNA to uncover signals of natural selection in Europe lost due to admixture or drift is now officially out in Nature Communications. In this paper we perform a haplotype homozygosity selection scan in aDNA from Europe.