Congrats to Peter, Ricky, and Mariana!
Congratulations to Peter for winning a departmental travel award, and to Ricky and Mariana for winning Dissertation Year Awards from UCLA!

Congratulations to Peter for winning a departmental travel award, and to Ricky and Mariana for winning Dissertation Year Awards from UCLA!

We are excited to work with Alexandria Hunt and Evelyn Barajas this summer, both Bruins in Genomics students!
Alexandria is an undergraduate student at UCLA in Computational and Systems Biology, and Evelyn recently graduated from Heritage University in Toppenish, WA with a B.S.
in Computer Science.


Left: Evelyn, Right: Alexandria
Richard Wolff recently presented his paper at the Microbiome Virtual International Forum and received an award for best talk in his session!
See the talk here:

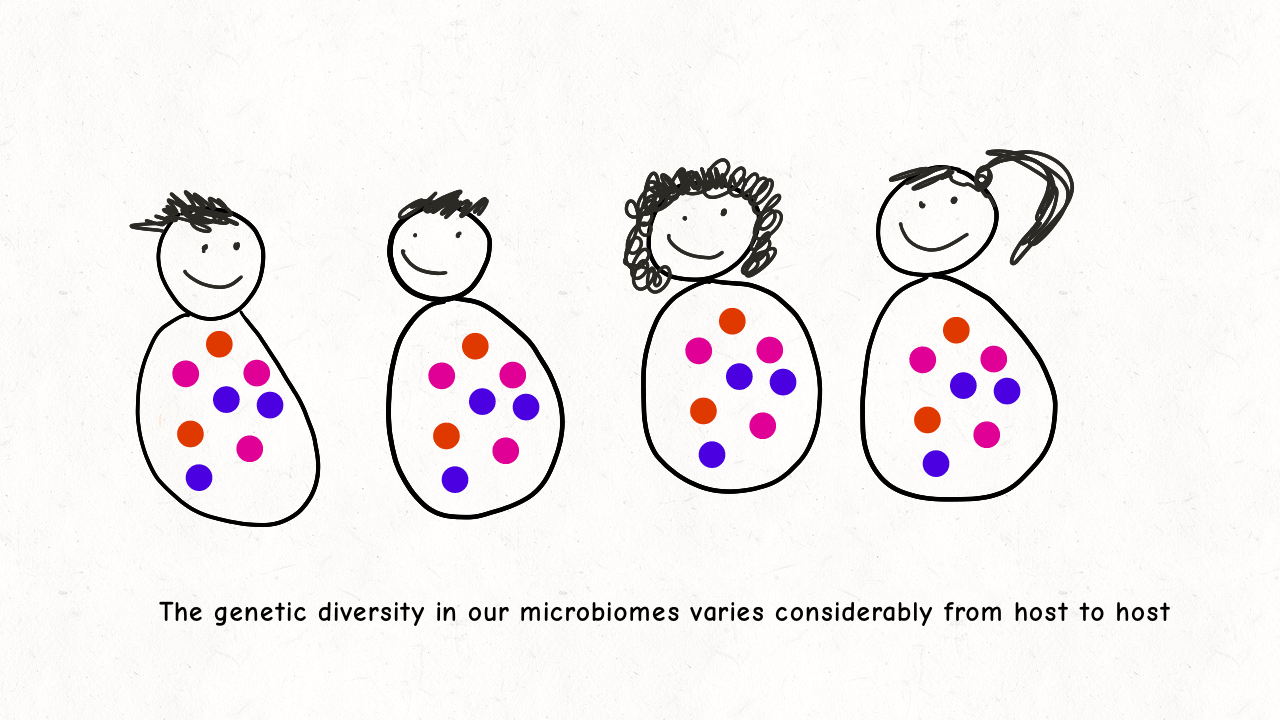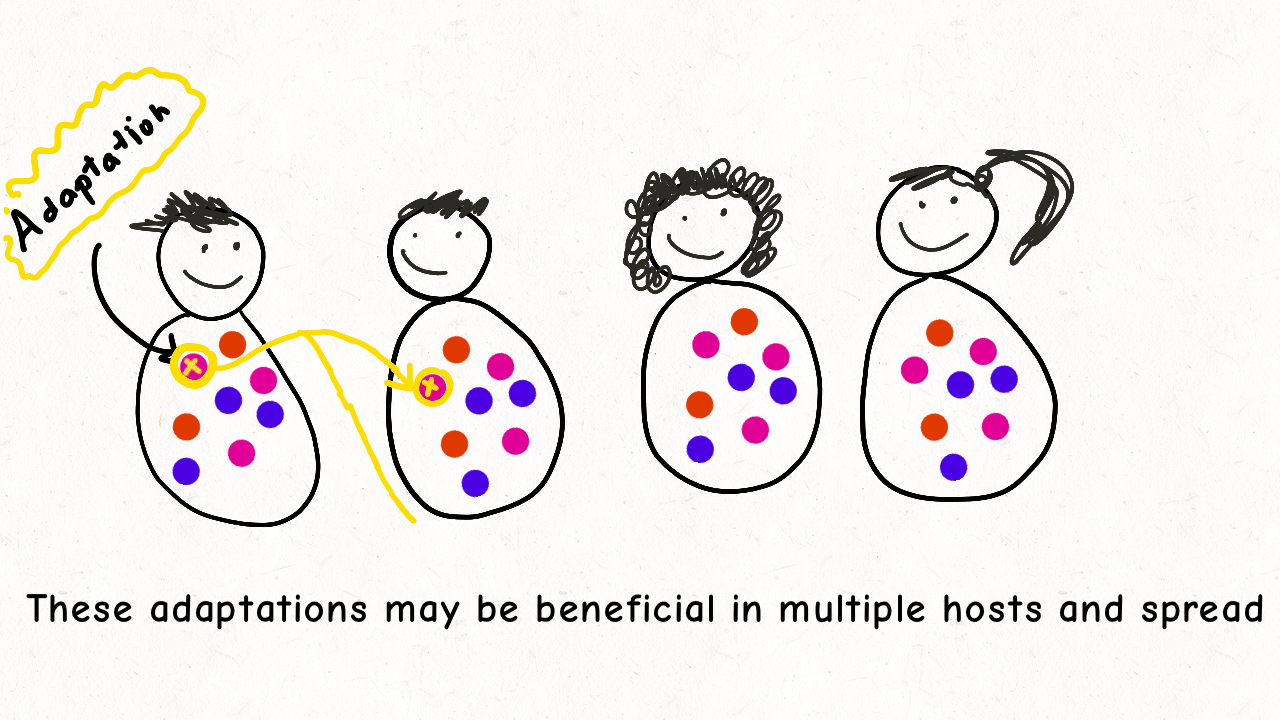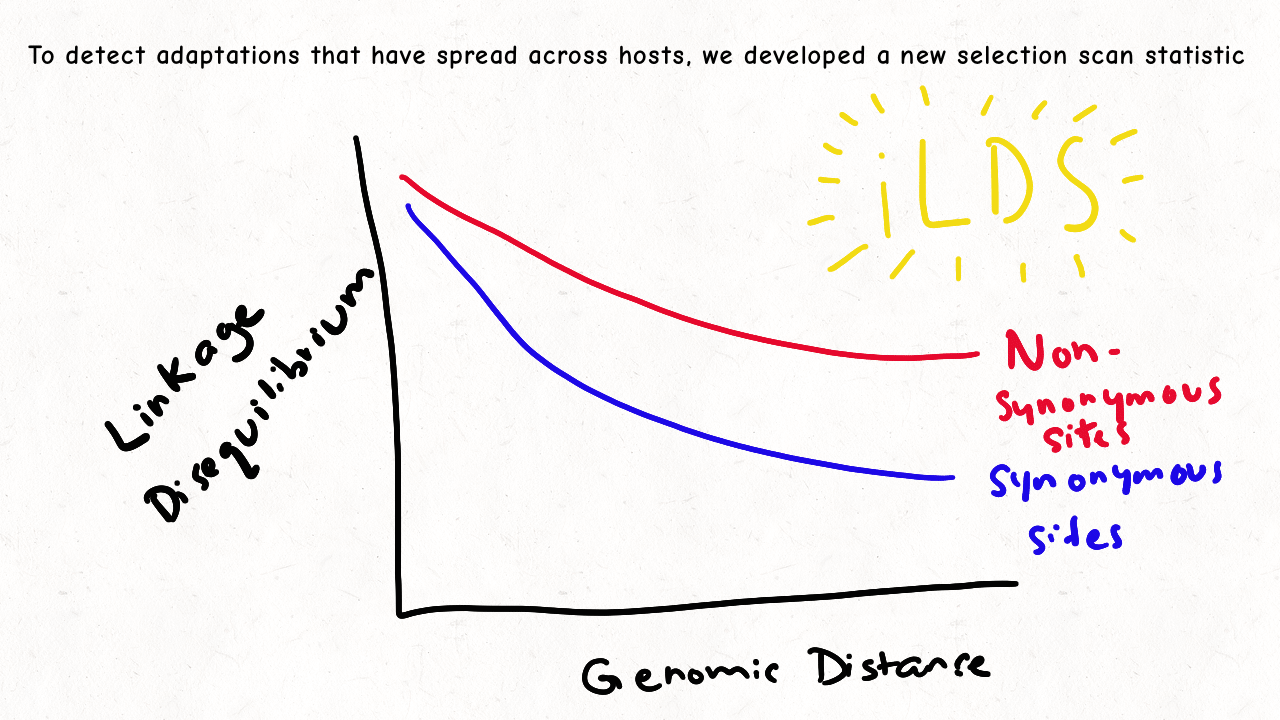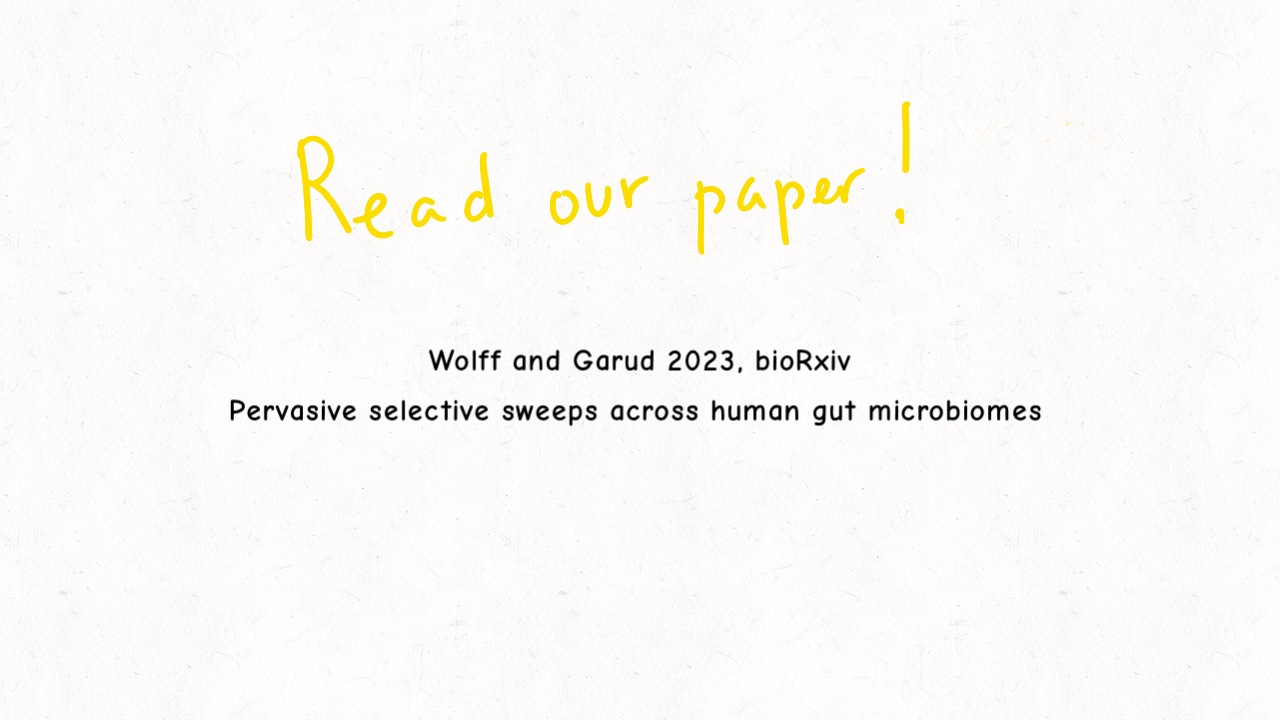
Richard Wolff and I are delighted to share our latest preprint entitled “Pervasive selective sweeps across human gut microbiomes.” Please check out our paper and our animation featuring the paper below!
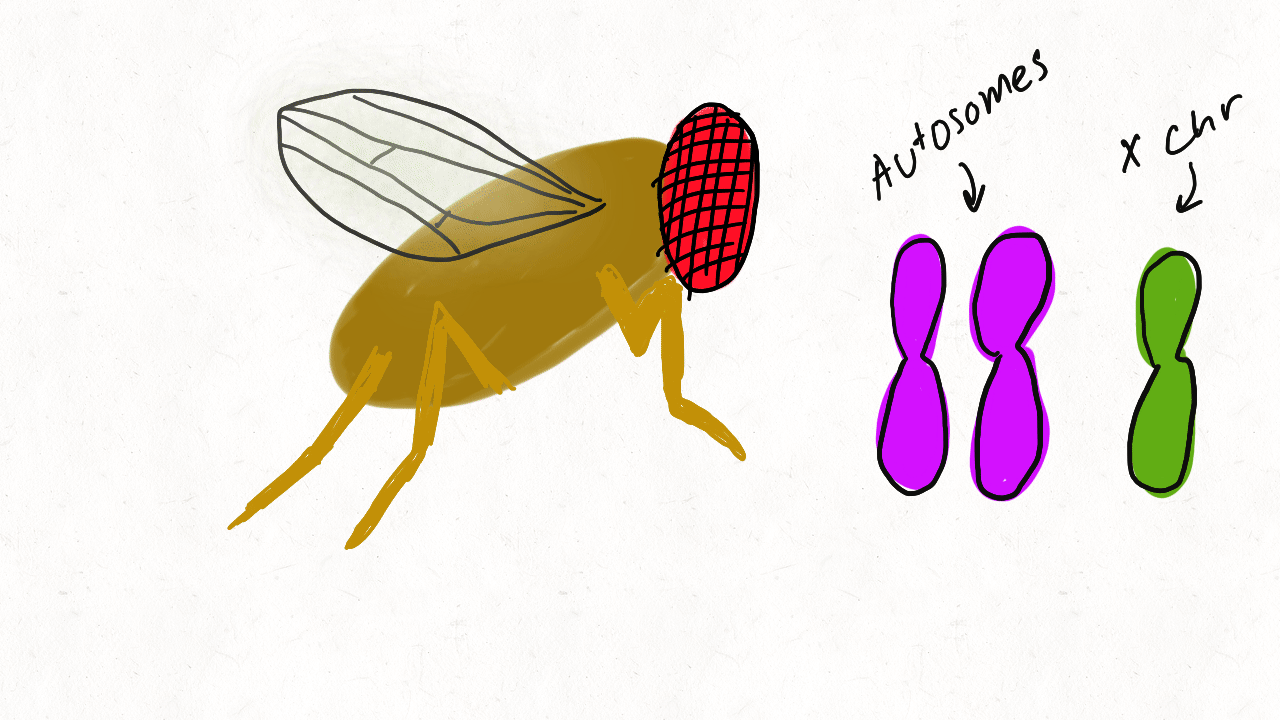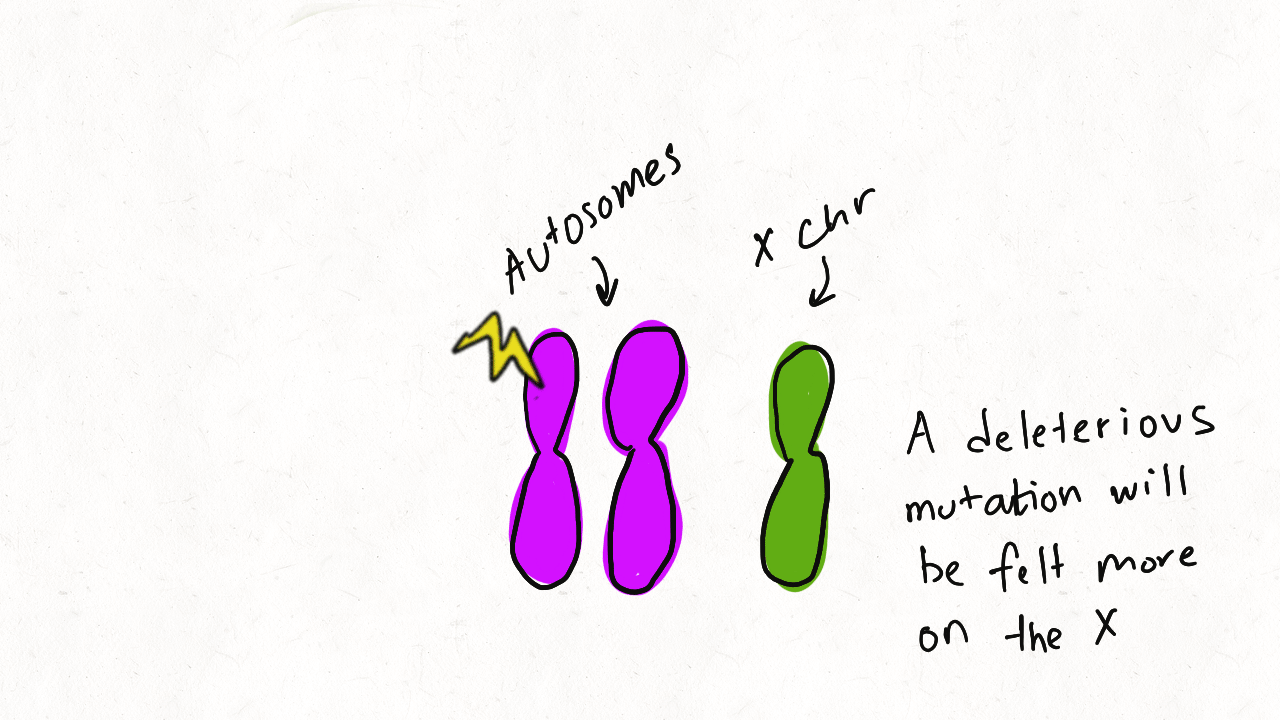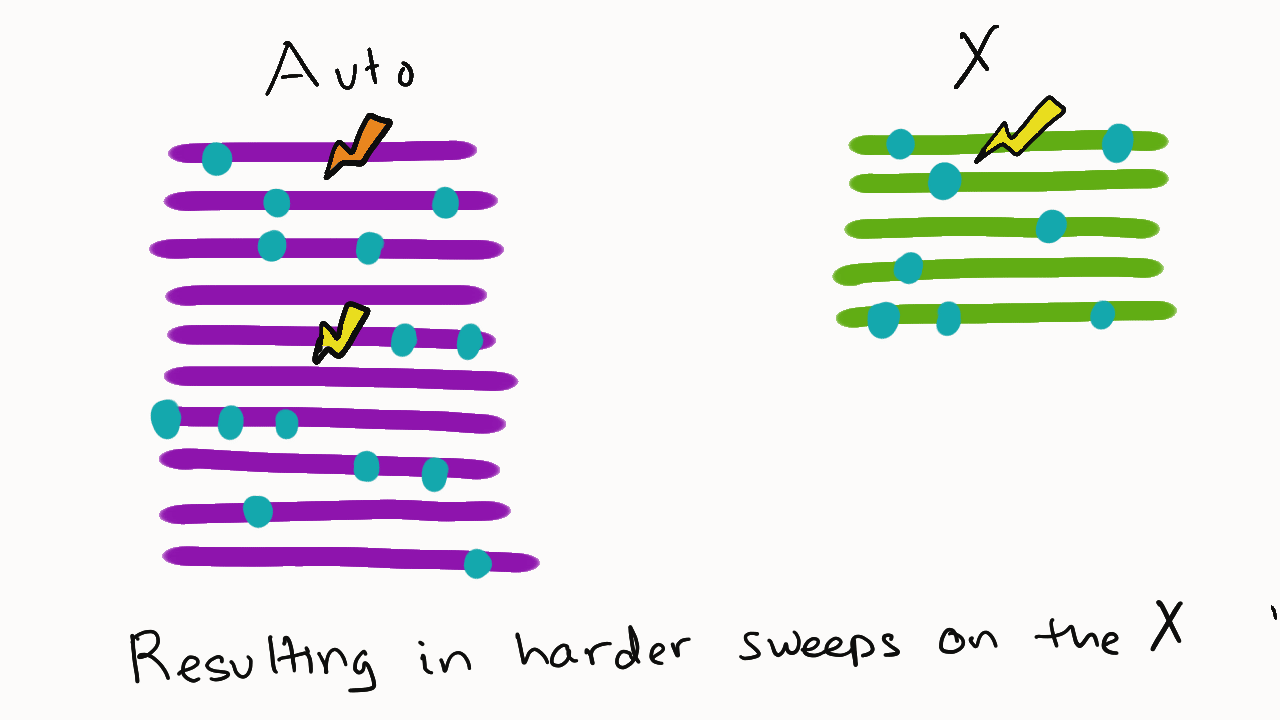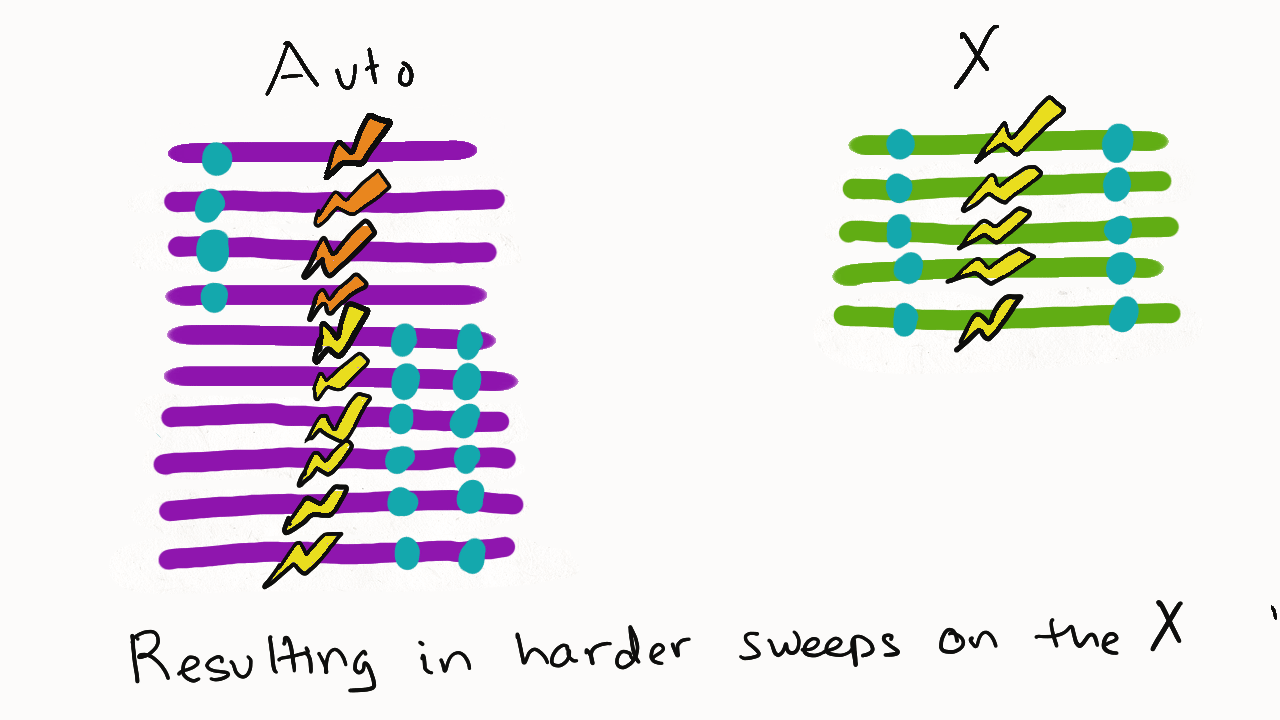
We are so proud of Mariana Harris for winning the DeLill Nasser Award! She will be attending PEQG this Spring with the award to present her work on the prevalence of hard versus soft sweeps on the X chromosome of Drosophila.
See her publications in Genetics (2024) and MBE (2023)!

We are incredibly proud of doctoral student Michael Wasney for winning the inaugural UCLA Goodman-Luskin Microbiome Center seed fellowship!


We are delighted to share our latest preprint on “Inference of the demographic histories and selective effects of human gut commensal microbiota over the course of human history“. Please check out our paper and below is an animation we made sharing a few highlights from the paper!


Congratulations to Mariana Harris for winning Best Poster Award at the UCLA QCBio retreat on her work on hard sweep enrichment on the X chromosomes of Drosophila melanogaster! Additionally, congratulations to Jonathan Mah for delivering a fantastic talk at the retreat on his work on demographic inference of gut commensal microbes!

We are extremely grateful to the NIH for awarding our lab an R35 grant. We will be working on furthering our understanding the evolutionary dynamics of gut bacteria during gut colonization. Stay tuned for results on this work!
Lead by Mariana Harris and in collaboration with Bernard Kim, we recently posted a preprint showing that hard sweeps are enriched on the X chromosome of six Drosophila species, generalizing findings that we previously made in a single population of D. melanogaster.
Please check out our video on the paper!