Microbial Evolution: An overlooked biomarker of host diet
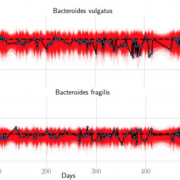
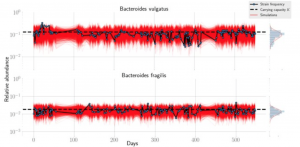
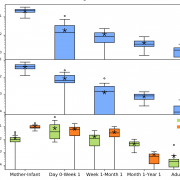
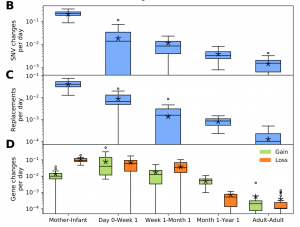
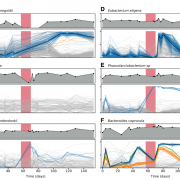
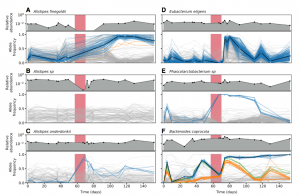
Nandita recently had the pleasure of writing a persective on a recent paper by Dapa et al. on the evolutionary dynamics of gut microbiota in response to diet. Dapa et al’s work highlights the importance of studying evolution of the gut microbiome to better understand how phenotypes impact our microbiota and vice versa. Remarkably, the authors find that diet is a driver of evolutionary change, and, remarkably, nucleotide-level signatures are a biomarker of host phenotype.
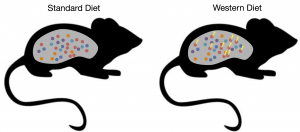
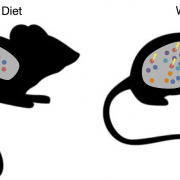
Adaptation of gut microbiota in response to diet B. thetaiotaomicron (red circles) evolves new genetic adaptations (yellow thunderbolts) in the presence of a high-fat and low-fiber Western diet.