Welcome BIG students!
This summer we are delighted to welcome four new BIG (Bruins in Genomics) summer interns to our group: Shavonna Jackson, Etan Dieppa, Kevin Delao, and Maya Singh!

Etan Dieppa

Maya Singh

Shavonna Jackson

Kevin Delao

This summer we are delighted to welcome four new BIG (Bruins in Genomics) summer interns to our group: Shavonna Jackson, Etan Dieppa, Kevin Delao, and Maya Singh!
Etan Dieppa
Maya Singh
Shavonna Jackson
Kevin Delao
Check out our latest paper on BioRxiv: https://www.biorxiv.org/content/10.1101/2020.06.20.163261v1
Whether hard sweeps or soft sweeps dominate adaptation has been a matter of much debate. Recently, we developed haplotype homozygosity statistics that (i) can detect both hard and soft sweeps with similar power and (ii) can classify the detected sweeps as hard or soft. The application of our method to population genomic data from a natural population of Drosophila melanogaster (DGRP) allowed us to rediscover three known cases of adaptation at the loci Ace, Cyp6g1, and CHKov1 known to be driven by soft sweeps, and detected additional candidate loci for recent and strong sweeps. Surprisingly, all of the top 50 candidates showed patterns much more consistent with soft rather than hard sweeps. Recently, Harris et al. 2018 criticized this work, suggesting that all the candidate loci detected by our haplotype statistics, including the positive controls, are unlikely to be sweeps at all and instead these haplotype patterns can be more easily explained by complex neutral demographic models. They also claim, confusingly, that these neutral non-sweeps are likely to be hard instead of soft sweeps. Here, we reanalyze the DGRP data using a range of complex admixture demographic models and reconfirm our original published results suggesting that the majority of recent and strong sweeps in D. melanogaster are first likely to be true sweeps, and second, that they do appear to be soft. Furthermore, we discuss ways to take this work forward given that the demographic models employed in such analyses are generally necessarily too simple to capture the full demographic complexity, while more realistic models are unlikely to be inferred correctly because they require fitting a very large number of free parameters.
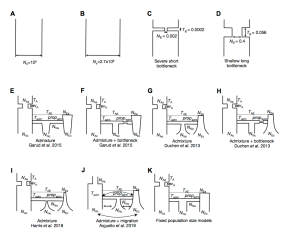
Demographic models tested in this paper
Today Leah Briscoe will speak about her work on batch correction of microbiome data for improved phenotype prediction at the QCBio seminar series!

Nandita had the opportunity to join the Women in Science group at USC for a virtual coffee hour where she and others had a chance to brainstorm ideas for working at home during the COVID-19 quarantine.
Some resources if you want to work on your computational chops at home:
CS50: Introduction to Computer Science (Harvard)
CS229: Machine Learning (Stanford)
Learning Bioinformatics at Home (Harvard)
The Rosalind Project (Bioinformatics)
SLiM self-guided tutorial (population genetics simulation software)
Writing Rocks: a daily routine for writing
We are delighted to welcome William Shoemaker and Ricky Wolff to our growing group!
Will is an NSF-funded postdoctoral scholar and joining us from his PhD lab in Indiana University.

Ricky is an MS student in the EEB department, recently graduated from Columbia University.

Thrilled to have you both join us!
Our latest collaboration on studying the Genetic Adaptation in NYC Rats is now on BioRxiv! Here we apply the G12 statistic, which can detect hard and soft sweeps from unphased data, to 29 whole genomes from NYC rats. We find intriguing signals of adaptation to urbanized environments in this population. Check out this perspective published in Nature on our work!
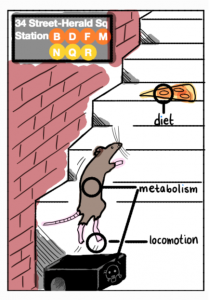
Figure replicated from our paper
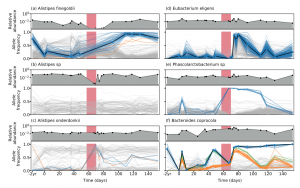
We are happy to share our latest collaboration examining the evolutionary dynamics in a single indivudual with high-res longitudinal data generated with long-read sequencing technology. Here, we show that genetic compositons within species can change in response to antibiotic perturbations even when the species relative abundance does not change significantly with time (see figure from the paper below). This indicates that the genetic changes observed within microbiomes play an important role in responding to selective pressures.
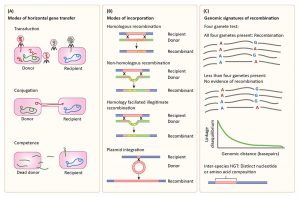
We are very happy that our review on Population Genetics in the Microbiome has been recently accepted to Trends in Genetics! The review will be officially published in January, but in the meanwhile, a copy is available here. Below is one of our figures illustrating modes of horizontal gene transfer, incorporation of DNA into the receipient’s genome, and statistical signatures of recombination.
We are excited to have Leah, Spencer, and Sara join us in the lab. Leah is a 3rd year PhD student working on developing new statistical methods to quanitfy ecological and evolutionary dynamics in microbial communities and is co-advised by Dr. Eran Halperin at UCLA. Sara is an MS student in EEB and Spencer is a 3rd year undergraduate student majoring in CS.
This quarter, Nandita will be teaching an EEB 297 course on Recombination in Bacteria. Please check out the syllabus with the list of readings and feel free to join us on Mondays from 2-4pm in 120 La Kretz Hall.