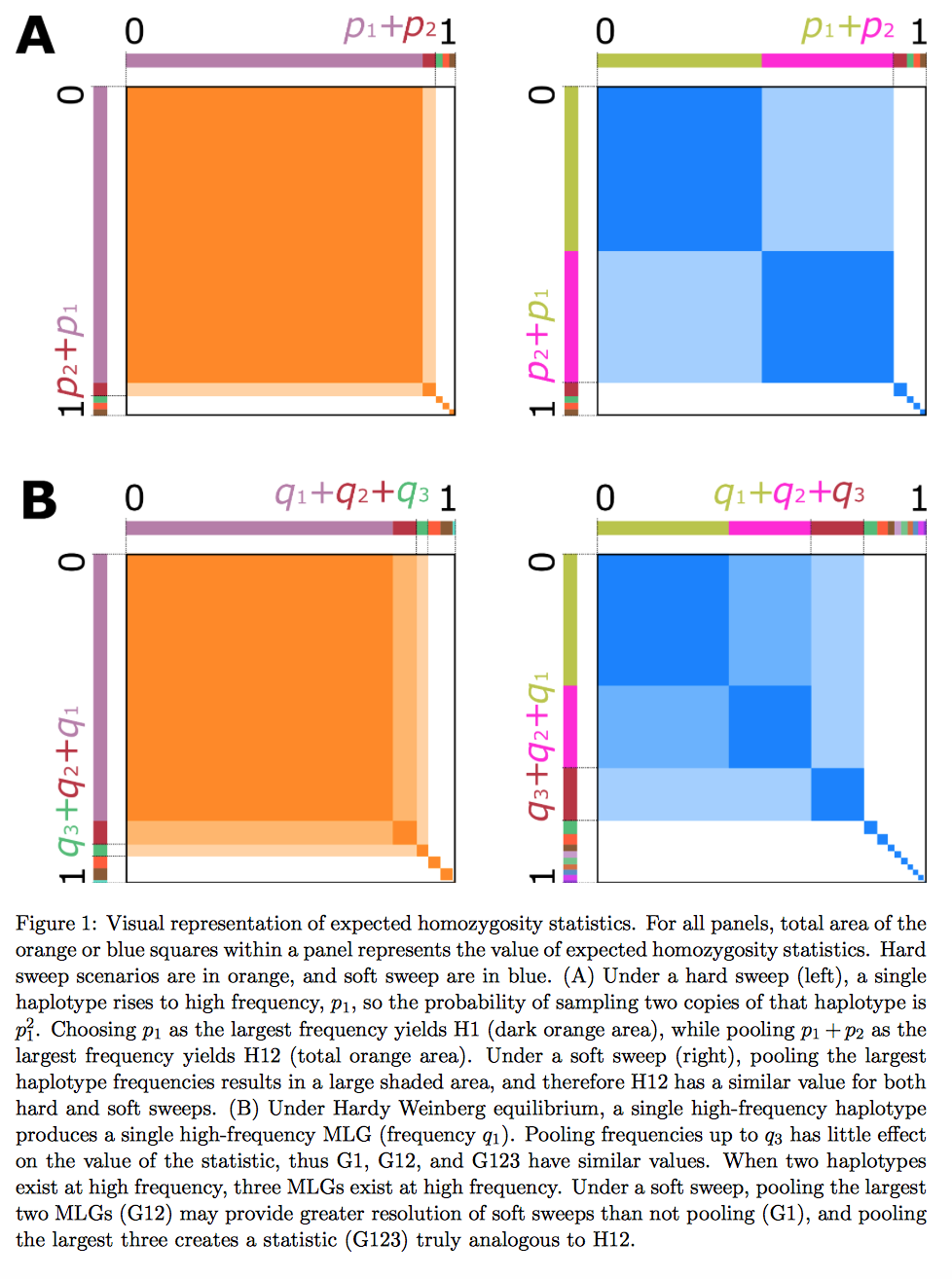
I am excited to share a new bioRxiv preprint on detecting and classifying hard and soft sweeps from unphased population genomic data, co-authored with Alexandre Harris and Michael DeGiorgio from Penn State University. One challenge with working with non-model eukaryotic organisms is that genomic data is often unphased, and therefore it is difficult to apply statistics intended for phased data. Here, we introduce the G12 and G123 statistics for detecting hard and soft sweeps from unphased data. These statistics are analogs to the H12 statistic for phased data (Garud et al. 2015). We also introduce G2/G1 to classify hard and soft sweeps analogously to H2/H1, conditional on a genomic region having high G12 and G123 values.
Please visit my github repository for code to compute H12, H123, H2/H1, G12, G123, and G2/G1:
Below: A visual depiction of H12 and G123.