
Welcome BIG summer students!
We are thrilled to welcome Jooeun Jeon, Radha Ganesh, and Rahul Natarajan this summer to our lab for the Bruins in Genomics summer program!

Radha Ganesh

Jooeun Jeon

Rahul Natarajan

We are thrilled to welcome Jooeun Jeon, Radha Ganesh, and Rahul Natarajan this summer to our lab for the Bruins in Genomics summer program!
Radha Ganesh
Jooeun Jeon
Rahul Natarajan

Our first PhD student, Leah Briscoe, defended her PhD this past month! Leah’s work spanned several topics, including background noise correction in the microbiome and source tracking in the microbiome with SNVs. She has one more paper coming out, please stay tuned! Congratulations Leah!


The lab celebrating Leah’s defense!

The Garud lab attended the Southern California Evolution meeting (SCALE) this past weekend and had a strong showing of presenters!

From left to right: Mariana, Michael, Aina, Peter, Ricky, and Jon

PhD student Mariana Harris gave a talk at SMBE Everywhere this month on her work on enrichment of hard sweeps on the X chromosome relative to autosomes. Congrats Mariana!

We are excited to share that our lab has received an NSF CAREER award to study rapid, repeatable adaptation in the human gut microbiome! Please stay tuned for results on this work!
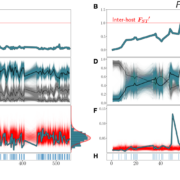
We are delighted to share that our paper entitled Ecological Stability Emerges at the Level of Strains in the Human Gut Microbiome is now accepted at mBio! This work is with the very talented first author and PhD student Richard Wolff and postdoc William Shoemaker.
In healthy hosts, species abundance fluctuations in the microbiome have been frequently described as stable, and these fluctuations can be described by macroecological laws. However, it is less clear how strain abundances change over time. An open question is whether individual strains behave like species themselves, exhibiting stability and following the macroecological relationships known to hold at the species level, or whether strains have different dynamics, perhaps due to the relatively close phylogenetic relatedness of co-colonizing lineages. Here, we analyze the daily dynamics of intra-specific genetic variation in the gut microbiomes of four healthy, densely longitudinally sampled hosts. First, we find that overall genetic diversity in a large majority of species is stationary over time, despite short-term fluctuations. Next, we show that fluctuations in abundances in approximately 80% of strains analyzed can be predicted with a stochastic logistic model (SLM)—an ecological model of a population experiencing environmental fluctuations around a fixed carrying capacity which has previously been shown to capture statistical properties of species abundance fluctuations. The success of this model indicates that strain abundances typically fluctuate around a fixed carrying capacity, suggesting that most strains are dynamically Finally, we find that the strain abundances follow several empirical macroecological laws known to hold at the species level. Together, our results suggest that macroecological properties of the human gut microbiome, including its stability, emerge at the level of strains.
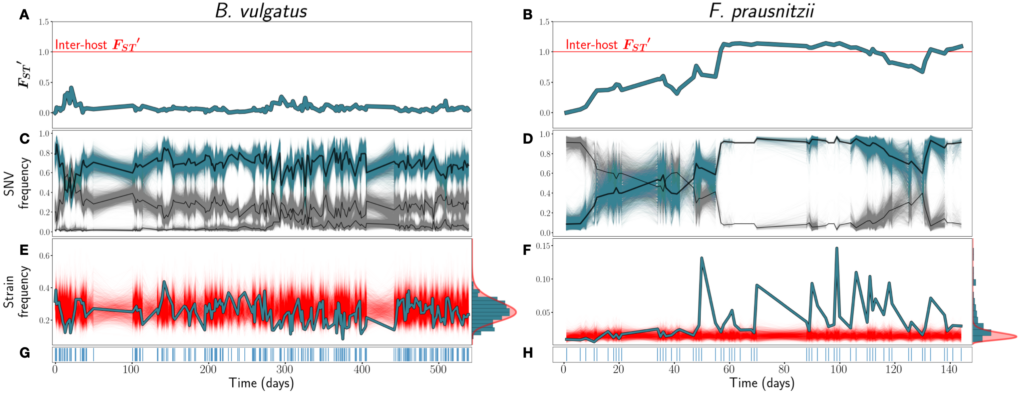
Figure from the paper: Figure 1: A & B) FST ′ trajectories for B. vulgatus (am) and F. prausnitzii (ao), respectively. C & D): SNV frequencies of three inferred strains for B. vulgatus (C) and two inferred strains for F. prauznitzii (D). In black are the inferred strain trajectories. Highlighted in blue are example strains featured further in (E) and (F). E & F) Frequencies of the example strains in blue, with simulations of the corresponding SLM overlaid in red. At right, the empirical distribution of strain abundances is plotted in blue, and the stationary Gamma distribution of abundances (see Equation 2) predicted by the SLM in red. G & H) Sampling time points. Blue lines indicate that a sample was taken on that day.
We are thrilled to share that our recent manuscript on the Enrichment of hard sweeps on the X chromosome of Drosophila melanogaster has now been accepted at Molecular Biology and Evolution! This work is with the very talented first author and PhD student, Mariana Harris.
The X chromosome is hemizygous in males and has ¾ Ne compared to autosomes. How do these factors impact the tempo and mode of adaptation (e.g. prevalence of hard versus soft sweeps) on the X compared to autosomes? Previously we showed with haplotype statistics (H12 and H2/H1) that soft sweeps are pervasive on autosomes in Drosophila melanogaster. These statistics since have been used in numerous other organisms to detect both hard and soft sweeps. https://tinyurl.com/soft-sweeps.
In this paper, we quantify the incidence of hard versus soft sweeps on the X chromosome with H12 and H2/H1 in both simulations and data. In simulations under a wide range of evolutionary scenarios, hard sweeps are expected to be more common than soft sweeps on the X. This arises from hemizygosity resulting in more efficient purging of initially deleterious standing variation, but also from the lower Ne on the X. Hard sweeps are enriched in D. mel data too. To apply H12 and H2/H1 to the data, we wanted to make sure that haplotype window sizes are comparable to that of autosomes. The X has fascinatingly lower levels of diversity compared to the autosomes, making comparable SNP-based window sizes based a little tricky. However, Mariana did some really great work in making this happen and I encourage you to read the paper to learn more.
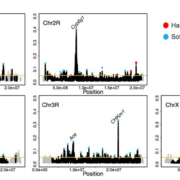
Here is a plot of the scan of H12 along all chromosomes – highlighted in blue are the inferred hard sweeps, and in red the inferred soft sweeps. The X has proportionally more hard sweeps than the auto! Also notice that we recovered a positive control, Fezzik, on the X with this scan, in addition to three positive controls on the auto at Ace, Cyp6g1, and CHKov1!

Figure from the paper: H12 scan in the DGRP data. H12 scan in DGRP data for four autosomal arms and the X chromosome. For the autosomal scan, each data point represents an H12 value in a 401-SNP window down sampled to 265 SNPs. For the X chromosome, windows of 265 SNPs were used. Regions with recombination rates <5 × 10−7 cM/bp were excluded from the scan and are denoted in gray. The golden line represents the 1-per-genome FDR line calculated under admixture Model 1 and a recombination rate of 5 × 10−7 cM/bp (see Materials and methods). The red and blue data points denote the top 50 and top 10 autosomal and X chromosome peaks, respectively. Blue and red data points correspond to the peaks that were classified as soft and hard sweeps, respectively, by our ABC approach with demographic Model 1, as described in the section “Softness of Sweeps on the X versus Autosomes”. The four positive controls (Ace, Cyp6g1, CHkov1, and Fezzik) are highlighted in the scan.
Finding that hard sweeps are more enriched the X in D. mel is the first step. How general is this phenomenon across diverse Drosophila species? This is an avenue for future work! Many thanks to our colleagues and anonymous reviewers for the very helpful feedback! We’d love to hear from you if you have any comments of questions about our paper. Also, Mariana will be at #Dros23!


Lab lunch in Westwood to celebrate the start of 2023!

Nandita had the pleasure of presenting a talk based on recent from the lab at a recent Keystone symposium on The Human Microbiome: Ecology and Evolution. PhD student, Richard Wolff also presented his poster on his paper on Ecological Diversity Emerges at the Level of Strains in the Human Gut Microbiome.



Garud Lab Halloween 2022, in front of the Life Sciences Building where we work.