
Farewell BIG students!
Thank you to Kaysa and Julianna for their hard work this summer!


Thank you to Kaysa and Julianna for their hard work this summer!

Garud lab picnic and bubble tea outings this summer:



We are extremely fortunate to have a stellar group of three PhD students joining the lab this year: Aina Martinez Zurita, Michael Wasney, and Peter Laurin. Welcome Aina, Michael and Peter!




We are trhilled to welcome our summer Bruins in Genomics interns!!!



We are thrilled to share that our paper entitled ‘Rapid evolution and strain turnover in the infant gut microbiome’ is now out in Genome Research! We find that the infant microbiome evolves faster than the adult microbiome, and moreoever, in our revision, we find that C-section infants have faster rates of evolution compared to vaginally-born infants. Please check out our paper!
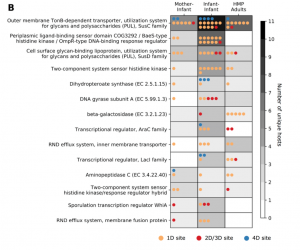
Figure 5b from our paper: We observe the same gene classes being mutated multiple times across infants and adults.

This was our first time attending a conference in-person as a group! Mariana, Jon, Ricky, and Nandita all presented posters on our work in the lab.



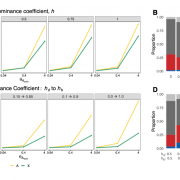
Recently Mariana Harris and Nandita Garud posted a preprint on hard and soft sweeps on the X versus autosome of D. melanogaster!
Here is the abstract!
The characteristic properties of the X chromosome, such as male hemizygosity and its unique inheritance pattern, expose it to natural selection in a way that can be different from the autosomes. Here, we investigate the differences in the tempo and mode of adaptation on the X chromosome and autosomes in a population of Drosophila melanogaster. Specifically, we test the hypothesis that due to hemizygosity and a lower effective population size on the X, the relative proportion of hard sweeps, which are expected when adaptation is gradual, compared to soft sweeps, which are expected when adaptation is rapid, is greater on the X than on the autosomes. We quantify the incidence of hard versus soft sweeps in North American D. melanogaster population genomic data with haplotype homozygosity statistics and find an enrichment of the proportion of hard versus soft sweeps on the X chromosome compared to the autosomes, confirming predictions we make from simulations. Understanding these differences may enable a deeper understanding of how important phenotypes arise as well as the impact of fundamental evolutionary parameters on adaptation, such as dominance, sex-specific selection, and sex-biased demography.
Please check out our work and let us know what you think!

Simulations suggest that sweeps on the X on average should be harder than on autosomes (Figure 2 of Harris and Garud, 2022)

Recently Leah Briscoe, Eran, Halperin, and Nandita Garud posted a preprint to bioRxiv on source tracking in the microbiome with single nucleotide variants.
In short, remember when this happened?

We wondered whether microbes could traverse oceans. To answer this question, we modified the input to a popular source tracking method called FEAST such that it takes in SNVs instead of speices. SNVs are known to provide higher resolution information than species about transmissions from sources to sinks, yet never have been incorporated in a source tracking algorithm. We found, surprisingly, that with SNVs but not speices there is a distance decay relationship between ocecans around the world!
We confirm the ability of SNVs over species in simualtions and applications to mother-infant data as well.
Please check out the paper and let us know what you think!
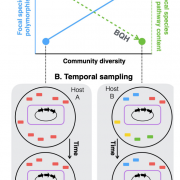
We are thrilled to share our latest preprint, Community diversity is associated with intra-species genetic diversity and gene loss in the human gut microbiome. This was a fun collaboration with Prof. Jesse Shapiro’s group in which we assessed the effect of community diversity on sub-species genetic diversity.
The human gut microbiome contains a diversity of microbial species that varies in composition over time and across individuals. These species are comprised of diverse strains, which are known to evolve by mutation and recombination within hosts. How the ecological process of community assembly interacts with sub-species diversity and evolutionary change is a longstanding question. Two hypotheses have been proposed based on ecological observations and theory: Diversity Begets Diversity (DBD), where taxa tend to become more diverse in already diverse communities, and Ecological Controls (EC), where higher community diversity impedes diversification within taxa. Recently we showed with 16S rRNA gene amplicon data from the Earth Microbiome Project that DBD is detectable in natural bacterial communities from a range of environments at high taxonomic levels (ranging from phylum to species-level), but that this positive relationship between community diversity and within-taxon diversity plateaus at high levels of community diversity. Whether increasing community diversity is associated with sub-species genetic diversity within microbiomes, however, is not yet known. To test the DBD and EC hypotheses at a finer genetic resolution, we analyzed sub-species strain and nucleotide variation in static and temporally sampled shotgun sequenced fecal metagenomes from a panel of healthy human hosts. We find that both sub-species single nucleotide variation and strain number are positively correlated with community diversity, supporting DBD. We also show that higher community diversity predicts gene loss in a focal species at a future time point and that community metabolic pathway richness is inversely correlated with the pathway richness of a focal species. These observations are consistent with the Black Queen Hypothesis, which posits that genes with functions provided by the community are less likely to be retained in a focal species’ genome. Together, our results show that DBD and Black Queen may operate simultaneously in the human gut microbiome, adding to a growing body of evidence that these eco-evolutionary processes are key drivers of biodiversity and ecosystem function.
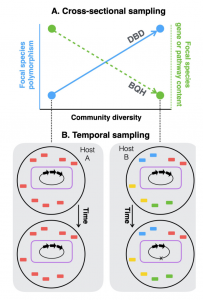
Figure 1 Diversity begets diversity(DBD) and the Black Queen Hypothesis(BQH) illustrated. See paper for more details!
We are thrilled to share that our paper on Evaluating supervised and unsupervised background noise correction in human gut microbiome data is finally out in PLoS Comp Bio! In this paper, we assess the ability of several noise correction methods to improve phenotype prediciton ability as well as biomarker discovery. We find utility in a Principal Component correction approach commonly used in other domains but to date has had seen limited application to microbiome data. Congrats to first-author Leah Briscoe!
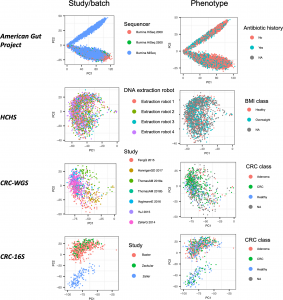
Figure 1 from our paper: Top PCs can sometimes correlate more with dataset label than disease. PCA applied to CLR-transformed taxonomic abundance data from the four datasets of the study. Each point represents a single microbiome sample colored by either study or batch and by phenotype group.