We are very proud to share our recent preprint:
Correcting for background noise improves phenotype prediction from human gut microbiome
Leah Briscoe, Bruna Balliu, Sriram Sankararaman, Eran Halperin and Nandita Garud
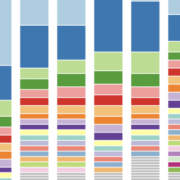
Many technical and biological variables can contribute to variation in microbiome data. For example, in this combined dataset, the study contributes more variation than colorectal disease status.

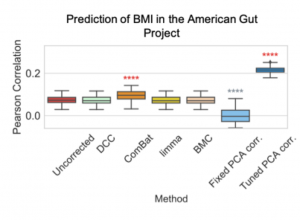
Many of the popular methods for correcting background noise in microbiome data are supervised. Unsupervised methods may be preferable because they can correct for unmeasured sources of variation. We perform a comparative analysis of background noise correction methods and find that regressing out Principal Components after applying a centered log-ratio transformation is quite effective in removing unwanted variation. In fact, if we correct for background noise, we can even improve phenotype prediction of many phenotypes.